Welcome to the Kastrup Lab. We are a group of biochemists and engineers working to gain a deeper understanding of how blood clotting is regulated and can be controlled therapeutically. Severe hemorrhage is a major cause of death, particularly among young people, while thrombosis is a major contributor to death and disability with age. We develop novel technologies for addressing unmet needs in controlling severe bleeding and preventing unwanted clotting. These technologies include RNA therapeutics, cellular therapies and other nanomedicines.
The research in our lab is focused on these questions:
- What new technologies are needed to halt bleeding in the most severe cases of hemorrhage?
- Can platelets be genetically modified to enhance their hemostatic activity, or targeted to treat Alzheimer’s disease?
- Can clot stability be altered to treat hemorrhage or thrombosis?
- Do soil and microbes in wounds influence bleeding?
We use a variety of biochemical assays, RNA biotechnology, biomaterial synthesis, imaging, patient blood, microfluidics and human blood, and small and large animal models to answer these questions.
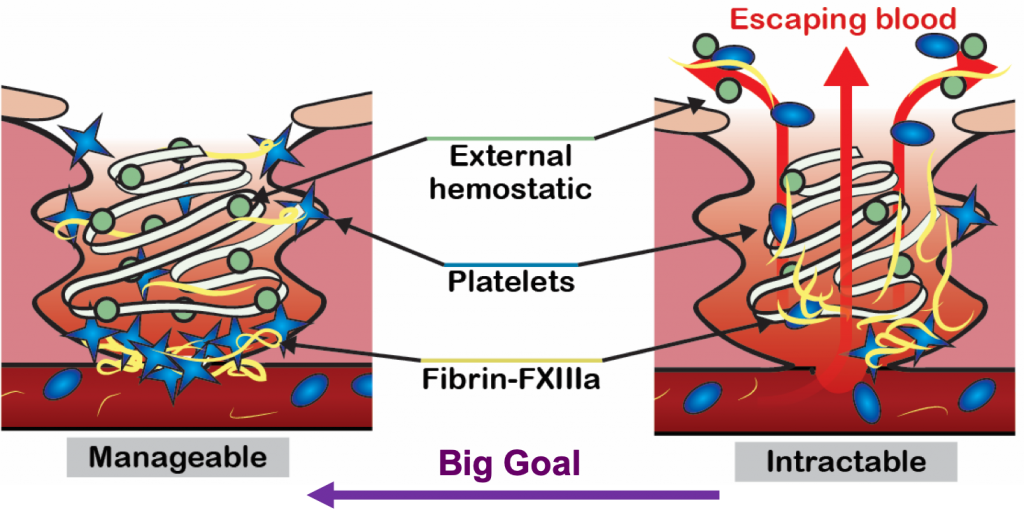 Severe hemorrhage can be triggered by trauma, surgery, or childbirth, and requires timely treatment with blood products and hemostatic agents to stop the blood loss. The most severe cases of hemorrhage are characterized by a widespread failure of the body’s blood clotting system, and bleeds can rapidly get out of control. Hemostatic agents are often applied topically into wounds to stop bleeding, but current agents are often ineffective in severe hemorrhage because they are flushed out of the wound by high blood flow.
Severe hemorrhage can be triggered by trauma, surgery, or childbirth, and requires timely treatment with blood products and hemostatic agents to stop the blood loss. The most severe cases of hemorrhage are characterized by a widespread failure of the body’s blood clotting system, and bleeds can rapidly get out of control. Hemostatic agents are often applied topically into wounds to stop bleeding, but current agents are often ineffective in severe hemorrhage because they are flushed out of the wound by high blood flow.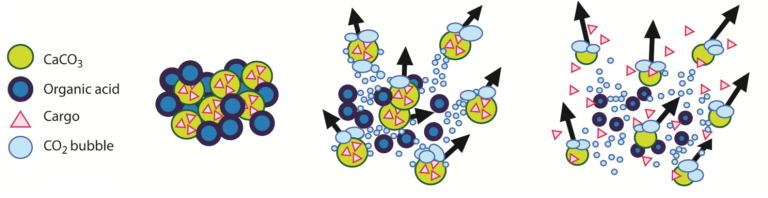 bleeding in trauma, surgery and endoscopy. These hemostatic products are being translated in collaboration with commercial and military partners. We are currently testing the ability of these microparticles to deliver antimicrobials into wounds to prevent infection during injury.
bleeding in trauma, surgery and endoscopy. These hemostatic products are being translated in collaboration with commercial and military partners. We are currently testing the ability of these microparticles to deliver antimicrobials into wounds to prevent infection during injury.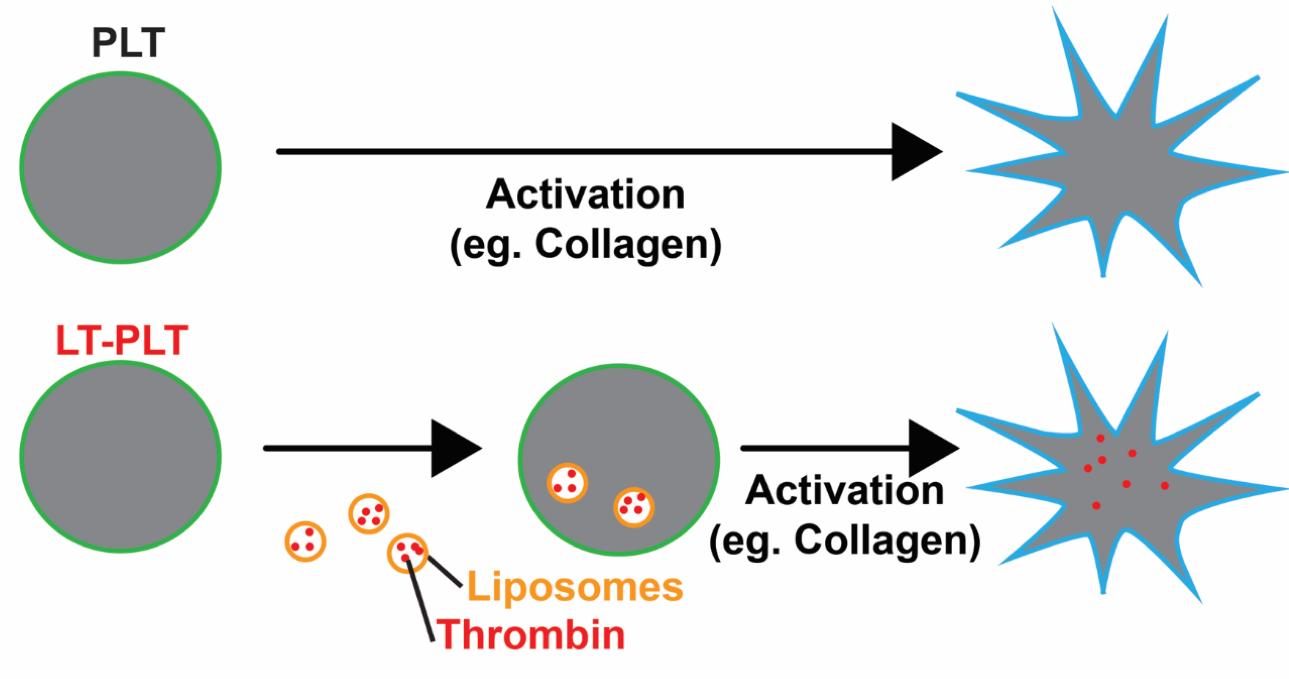 Platelets are small, anucleate blood cells essential for hemostasis. In response to vascular damage, platelets activate and aggregate to form a plug to seal the wound and prevent blood loss. As part of this process, activated platelets release molecules that promote clot formation in a localized manner.
Platelets are small, anucleate blood cells essential for hemostasis. In response to vascular damage, platelets activate and aggregate to form a plug to seal the wound and prevent blood loss. As part of this process, activated platelets release molecules that promote clot formation in a localized manner. 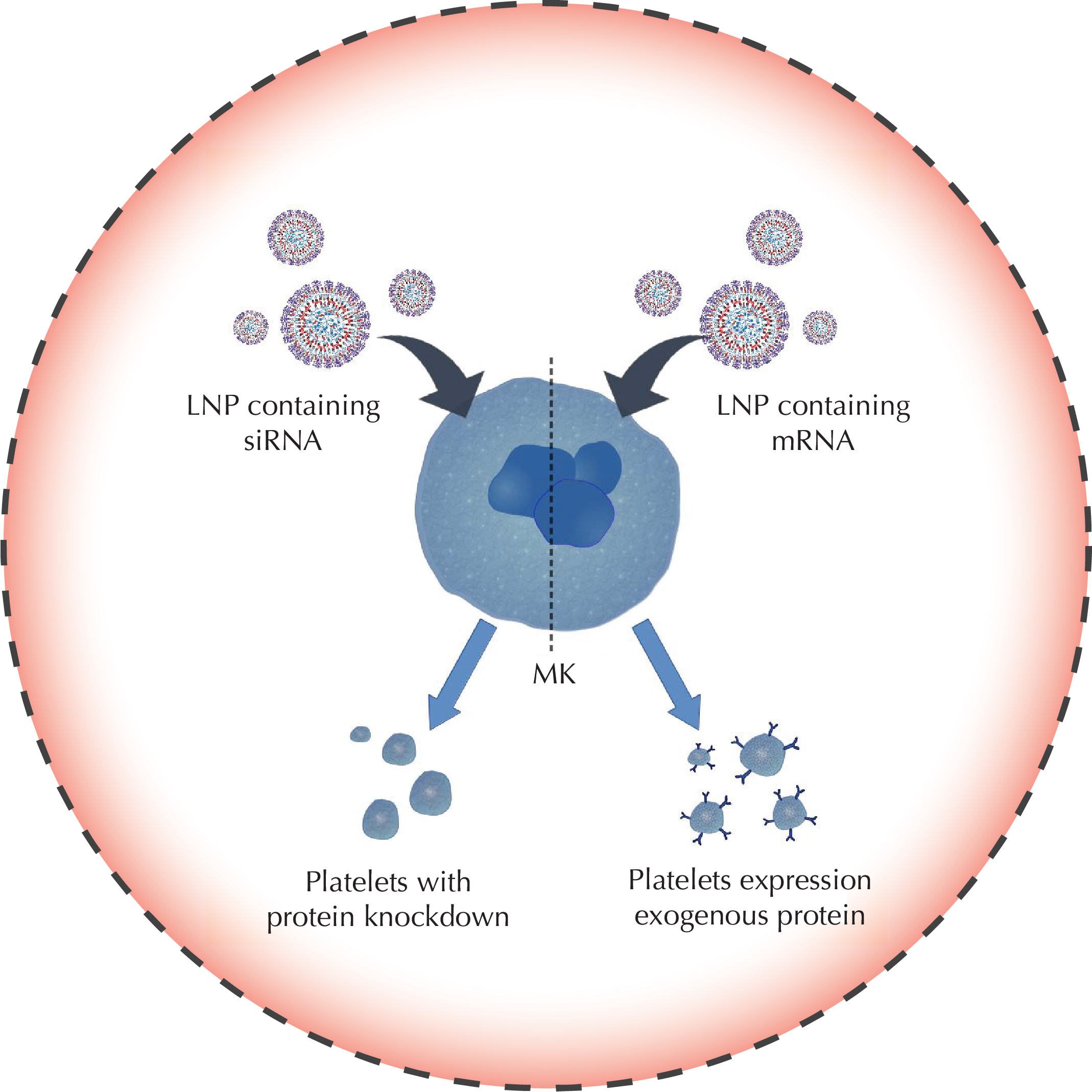
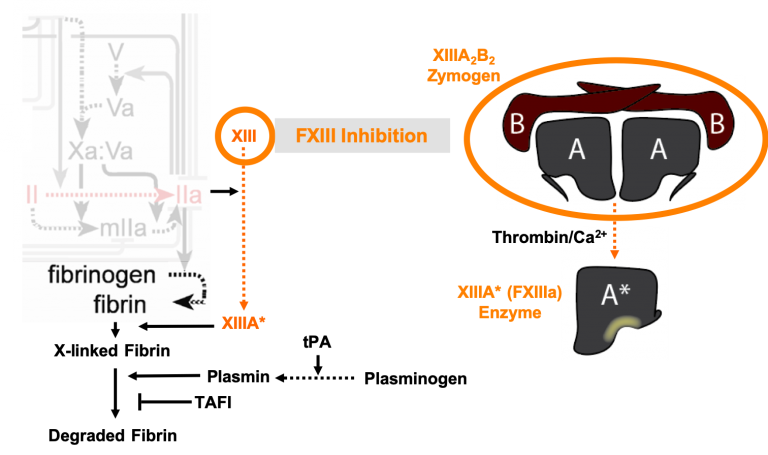 During hemorrhage, fibrin is covalently crosslinked to itself and the vessel wall to form a meshwork that seals the wound. If this meshwork is not stably linked to the vessel wall, the wound is not effectively sealed, leading to re-bleeding. This process is mediated by coagulation factor XIIIa (FXIIIa), a transglutaminase enzyme found in platelets and blood. We are currently exploring whether supplementing the blood with additional FXIIIa is a feasible strategy to strengthen clots that form during severe bleeding. Furthermore, our lab has shown that the enzymes responsible for degrading fibrin, tissue plasminogen activator (tPA) and plasmin, also degrade FXIIIa. Since tPA is used clinically as clot-busting drug in patients with thrombosis, we are investigating whether the activity of FXIIIa is altered in patients that have received this treatment.
During hemorrhage, fibrin is covalently crosslinked to itself and the vessel wall to form a meshwork that seals the wound. If this meshwork is not stably linked to the vessel wall, the wound is not effectively sealed, leading to re-bleeding. This process is mediated by coagulation factor XIIIa (FXIIIa), a transglutaminase enzyme found in platelets and blood. We are currently exploring whether supplementing the blood with additional FXIIIa is a feasible strategy to strengthen clots that form during severe bleeding. Furthermore, our lab has shown that the enzymes responsible for degrading fibrin, tissue plasminogen activator (tPA) and plasmin, also degrade FXIIIa. Since tPA is used clinically as clot-busting drug in patients with thrombosis, we are investigating whether the activity of FXIIIa is altered in patients that have received this treatment. Coagulation is controlled by a complex network of reactions that were first described over 50 years ago. Research over the decades has led to a better understanding of the biochemistry of the coagulation network, yet questions still remain about aspects of blood clotting. Humans deficient in coagulation factor XII (FXII) do not appear to bleed spontaneously or excessively after trauma, leading to a debate over its role in blood clotting. FXII is activated by silicates, a major component of soil, to clot blood
Coagulation is controlled by a complex network of reactions that were first described over 50 years ago. Research over the decades has led to a better understanding of the biochemistry of the coagulation network, yet questions still remain about aspects of blood clotting. Humans deficient in coagulation factor XII (FXII) do not appear to bleed spontaneously or excessively after trauma, leading to a debate over its role in blood clotting. FXII is activated by silicates, a major component of soil, to clot blood 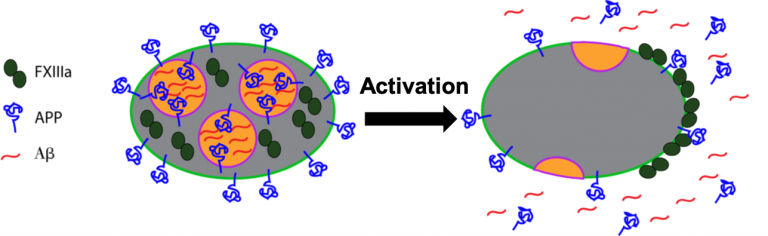 Neurodegenerative diseases have also been linked to defective clotting. For example, amyloid beta (Aβ), a key protein associated with Alzheimer’s disease, interacts with coagulation proteins and can accumulate along the inside of blood vessels during cerebral amyloid angiopathy. We recently showed that FXIIIa can crosslink Aβ to itself and to fibrinogen to make clots stiffer. We are currently exploring the physiological significance of this interaction to determine whether Aβ is a normal component of blood clots in the brain, or if FXIIIa contributes to the pathology of cerebral amyloid angiopathy. As platelets are an abundant source of Aβ precursor protein, we are also investigating the physiological role of Aβ precursor protein in platelets.
Neurodegenerative diseases have also been linked to defective clotting. For example, amyloid beta (Aβ), a key protein associated with Alzheimer’s disease, interacts with coagulation proteins and can accumulate along the inside of blood vessels during cerebral amyloid angiopathy. We recently showed that FXIIIa can crosslink Aβ to itself and to fibrinogen to make clots stiffer. We are currently exploring the physiological significance of this interaction to determine whether Aβ is a normal component of blood clots in the brain, or if FXIIIa contributes to the pathology of cerebral amyloid angiopathy. As platelets are an abundant source of Aβ precursor protein, we are also investigating the physiological role of Aβ precursor protein in platelets.
Recent Comments